Nat Communications | Structure of human NaV1.6 channel reveals Na+ selectivity and pore blockade by 4,9-anhydro-tetrodotoxin
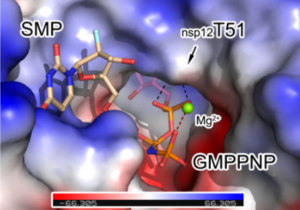
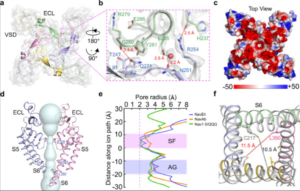
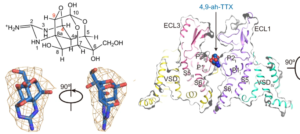
The sodium channel NaV1.6 is widely expressed in neurons of the central and peripheral nervous systems, which plays a critical role in regulating neuronal excitability. Dysfunction of NaV1.6 has been linked to epileptic encephalopathy, intellectual disability and movement disorders. Here we present cryo-EM structures of human NaV1.6/β1/β2 alone and complexed with a guanidinium neurotoxin 4,9-anhydro-tetrodotoxin (4,9-ah-TTX), revealing molecular mechanism of NaV1.6 inhibition by the blocker. The apo-form structure reveals two potential Na+ binding sites within the selectivity filter, suggesting a possible mechanism for Na+ selectivity and conductance. In the 4,9-ah-TTX bound structure, 4,9-ah-TTX binds to a pocket similar to the tetrodotoxin (TTX) binding site, which occupies the Na+ binding sites and completely blocks the channel. Molecular dynamics simulation results show that subtle conformational differences in the selectivity filter affect the affinity of TTX analogues. Taken together, our results provide important insights into NaV1.6 structure, ion conductance, and inhibition.